Sweet Adaptation
Preschoolers should spend their time running, jumping and improving their fine motor skills, but a small number of children experience the opposite. Instead, they slowly lose the skills they have only recently acquired and begin to lose feeling in their arms and legs. As this condition progresses, they become quadriplegics, eventually becoming dependent on feeding tubes and ventilators before they die, typically in their 20s or 30s. This devastating disease, called Giant Axonal Neuropathy (GAN), has fewer than 100 known cases, which makes research – and therefore treatment options – scarce. A collaboration between Center for Genomic and Computational Biology (GCB) member Ashley Chi, Ph.D. and assistant professor of biochemistry, Michael Boyce is shedding some new light on this disease, and their efforts may also help treat a variety of other neurological diseases and cancers.
GAN is a recessively inherited condition – both parents must carry one copy of the mutated GAN gene – that results in progressive nerve death. The GAN gene provides instructions for making the adaptor protein gigaxonin, which belongs in the Kelch-like (KLHL) family of proteins. KLHL proteins are involved in a cellular function that destroys and removes excess or damaged proteins using a mechanism called the ubiquitin proteasome system.
“The Kelch family of proteins acts like a garbage collector, but what happens when the garbage workers go on strike?,” Chi said. “ You can’t get rid of your garbage, so you end up storing all of it, and it creates issues.”
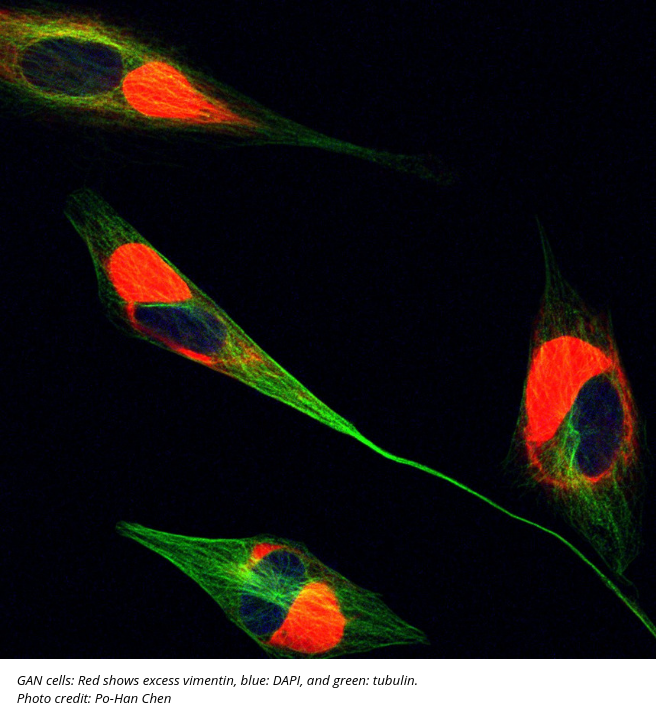
When neurons in GAN patients aren’t able to process excess proteins like vimentin and neurofilaments, they can accumulate in the axons of nerve cells and enlarge the size of axon, hence the name “Giant Axonal Neuropathy.” These accumulated proteins eventually impair the function of neurons and cause complications like loss of feeling and mobility, and, characteristically in patients with GAN, an excess of tightly curled hair.
Chi and Boyce, received a pilot award from the Hannah’s Hope Fund, a GAN-focused private foundation, and, more recently, an R01 grant from the National Institute of Neurological Disorders and Stroke (NINDS) for $2.12 million to investigate the importance of KLHL proteins like gigaxonin and how tweaking the mechanism behind them can affect the backlog of proteins in the cells. This could provide new treatment options for patients with GAN and other diseases.
This is the first time researchers have established that the adaptor protein is responsible for regulating the metabolism and nutrient status of the cells in this way. “People used to think that the adaptor protein was no big deal,” Chi said, “but now we are seeing that it is not only interesting but also important to cellular function.”
The team found a sugar modification, O-linked-β-linked N-acetylglucosamine (O-GlcNAc), that may help eliminate the backlog of proteins. Like eating a cookie and leaving a crumb behind, cells take glucose from the outside and leave a “crumb” of sugar behind on the protein. If a cell has a lot of glucose, the adaptor protein gets decorative sugar; a low level of glucose, no decorative sugar. Po-Han Chen, Ph.D., a former Duke graduate student co-mentored by Chi and Boyce, found that without sugar, KLHL proteins like gigaxonin are not functional. These results, which may have implications for diseases ranging from cancer to neurodegeneration, were published in an initial paper in The Embo Journal; a second paper is currently under review.
With the support of Hannah’s Hope Fund and NINDS, Chi and Boyce will conduct studies using both mouse models and CRISPR-edited GAN cells in which they will add sugar to mutated KLHL adaptor proteins to regulate them and improve their function. They hope these experiments will open the door to new therapeutic opportunities to treat GAN, other neurodegenerative diseases and cancer.
Finding new treatment paths may help those affected by GAN reclaim their lives in the future so toddlers can keep on toddling and parents can watch their children grow up well into adulthood without needing braces, feeding tubes and ventilators.
In addition to the HHF and NINDS grants, this project has been supported by pilot funding from the Duke Cancer Institute and the Duke O'Brien Center for Kidney Research.